
Qu’est-ce que la maladie de Huntington ?
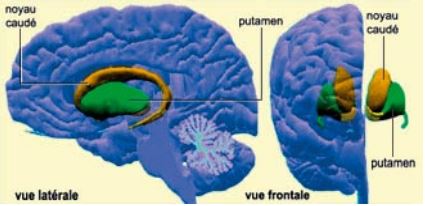
La maladie de Huntington, également dénommée chorée de Huntington, est une affection génétique et héréditaire conduisant à la destruction des neurones de certaines régions cérébrales. Elle se traduit principalement par des mouvements anormaux et des troubles du comportement. C’est une affection neurodégénérative touchant surtout le striatum (structure située au milieu du cerveau) : noyau caudé et putamen, et ultérieurement le cortex cérébral.
On connaît la maladie depuis plus d’une centaine d’années puisque le docteur George Huntington l’a décrite en 1872 mais le gène responsable n’a été découvert qu’en 1993.
Combien de personnes sont atteintes de la maladie ?
La maladie atteint environ une personne sur 10 000 en France, ce qui représente 6000 malades et 12 000 porteurs du gène provisoirement indemnes de signes cliniques.
Qui peut en être atteint ?
Elle frappe le plus souvent des personnes entre 30 et 45 ans, sans prédominance de sexe ni d’ethnie. Elle est cependant plus fréquente dans les populations d’origine européenne et peut parfois survenir aux deux extrêmes de la vie, soit chez des enfants soit chez des personnes âgées de plus de 70 ans.
Est-elle contagieuse ?
Non, les maladies génétiques ne sont pas contagieuses.
Quelles en sont les manifestations ?
Il y a trois types de symptômes principaux dans la maladie de Huntington :
– les troubles moteurs avec une maladresse, des mouvements anormaux involontaires (chorée), des troubles de la posture et de l’équilibre entraînant une gêne à la marche et des diffi cultés à articuler puis à déglutir.
– les troubles du comportement avec une modifi cation de la personnalité, une tendance à la dépression, ou au contraire plus rarement un état maniaque avec excitation, hyperactivité, irritabilité, des troubles obsessionnels, un état psychotique, voire même des conduites agressives vis-à-vis de soi-même et/ou vis-à-vis d’autrui.
– Les troubles cognitifs avec une perte de mémoire, des diffi cultés de concentration, d’abstraction, des erreurs de jugement, une désorientation dans l’espace, des diffi cultés d’organisation et d’apprentissage, des problèmes pour s’adapter au changement… Il existe aussi des diffi cultés à interpréter les relations avec autrui ce qui peut causer des malentendus au quotidien.
Il faut mettre un peu à part les formes de l’enfant qui se traduisent par des symptômes différents. Les enfants atteints ont des gestes lents et des raideurs ; ils éprouvent des diffi cultés d’apprentissage et peuvent avoir des convulsions. Certains ont de graves anomalies
du comportement.
Quelle est son évolution ?
Au début de la maladie, les mouvements anormaux apparaissent souvent au premier plan mais les troubles du comportement ainsi que des troubles des fonctions cognitives peuvent être présents d’emblée.
La maladie évolue sur de nombreuses années et conduit progressivement à une perte d’autonomie, puis fi nalement au décès au bout de quinze à vingt ans en moyenne, sachant que l’évolution de chaque patient reste un cas particulier et est donc impossible à prédire. L’évolution est d’autant plus rapide que la maladie survient tôt dans la vie. Les formes de l’enfant, notamment, ont une évolution plus rapide.
Comment expliquer les symptômes ?
Cette maladie est d’origine génétique. Les chromosomes, support du patrimoine génétique, sont composés d’ADN et de protéines. Les gènes sont des fragments d’ADN. Chaque gène, lorsqu’il est connu et identifi é, est localisé précisément sur un chromosome. La maladie de Huntington est due à une anomalie (mutation) d’un gène nommé IT15 et situé sur le bras court du chromosome 4. La molécule d’ADN est constituée de quatre bases (qui constituent l’alphabet du code génétique), à savoir A(adénine), T(thymine), G(guanine), et C(cytosine). Le gène responsable de la maladie de Huntington possède une région dans laquelle une séquence de trois bases (CAG) est répétée de nombreuses fois. Le gène muté comporte une augmentation du nombre de ces répétitions. Ce gène porte l’information pour la fabrication d’une protéine, la huntingtine, dont la fonction normale est inconnue à ce jour, même si l’on sait qu’elle a un rôle protecteur sur le cerveau. La mutation responsable rend toxique la protéine huntingtine mutée. Celle-ci forme des agrégats dans le noyau des neurones du noyau caudé, puis du cortex cérébral. Il reste à établir le mécanisme exact de la toxicité de la protéine anormale. Dans un faible pourcentage de cas, la maladie de Huntington n’est pas liée à des anomalies de IT15. D’autres gènes peuvent alors être en cause.